
Únase a nosotros el 30 de abril: Presentación de la prueba CT de Parasoft C/C++ para pruebas continuas y excelencia en el cumplimiento | Regístrese ahora
Saltar a la sección
Navegando por la guía de validación de la FDA para la ciberseguridad de dispositivos médicos

13 de noviembre.
13 min leer
Con la adición de más requisitos por parte de la FDA a sus estándares y validación de ciberseguridad, surge la necesidad de que los fabricantes de software médico adopten análisis estáticos para garantizar que su software cumpla con estos nuevos estándares de seguridad. Continúe leyendo para aprender cómo implementar análisis estático para cumplir con estos requisitos de seguridad.
Saltar a la sección
Saltar a la sección
A medida que la FDA agrega más requisitos de ciberseguridad a su guía de validación de software, los fabricantes de dispositivos médicos pueden recurrir al análisis estático, el método más eficaz para abordar los problemas de seguridad y ofrecer software predecible.
Los fabricantes de dispositivos médicos continúan centrándose en mejorar los procesos de desarrollo de software por dos razones principales.
- Abordar las crecientes amenazas a la seguridad.
- Satisfacer los requisitos de la FDA, que son cada vez más precisos.
Comprensión de la nueva guía de ciberseguridad de la FDA
La nueva guía de ciberseguridad para dispositivos médicos de la FDA se publicó en septiembre de 2023 y proporciona recomendaciones integrales a la industria con respecto al diseño, el etiquetado y la documentación de los dispositivos de ciberseguridad que se incluirán en las presentaciones previas a la comercialización de dispositivos con riesgo de ciberseguridad.
La guía tiene como objetivo ayudar a los fabricantes a identificar y mitigar los riesgos de ciberseguridad a lo largo del ciclo de vida de un dispositivo médico, desde el diseño y el desarrollo hasta la fabricación, el servicio y la vigilancia posterior a la comercialización. También proporciona recomendaciones para comunicar los riesgos de ciberseguridad a los proveedores de atención médica y a los pacientes.
La nueva guía se basa en los documentos de orientación de ciberseguridad anteriores de la FDA, pero también incluye una serie de recomendaciones nuevas y actualizadas, como:
- Un requisito para que los fabricantes implementen un programa integral de gestión de riesgos de ciberseguridad.
- Un requisito para que los fabricantes presenten una lista de materiales de software (SBOM) para sus dispositivos.
- Un requisito para que los fabricantes desarrollen e implementen planes de ciberseguridad para el soporte poscomercialización.
- Recomendaciones para comunicar los riesgos de ciberseguridad a los proveedores de atención médica y a los pacientes.
La FDA solía centrarse en seguridad funcional aspectos de los sistemas, pero ahora la seguridad cibernética es un tema de igual importancia. Aunque la seguridad y la protección son similares (y fácilmente se podría argumentar que ambas tienen que ver con la creación de software predecible), la FDA considera la ciberseguridad algo que requiere atención y medidas dedicadas.
La importancia de la validación de procesos en dispositivos médicos
La validación de procesos es la recopilación y evaluación de datos que establecen evidencia científica de que un proceso es capaz de entregar consistentemente un producto de calidad. En el contexto de los dispositivos médicos, la validación de procesos es esencial para garantizar la seguridad y eficacia de los dispositivos durante todo su ciclo de vida.
La validación del proceso es particularmente importante para los dispositivos médicos controlados por software, ya que el software puede ser complejo y difícil de probar de manera integral. Por ejemplo, un dispositivo médico controlado por software puede tener millones de líneas de código y no es posible probar todos los escenarios y combinaciones posibles de entradas y salidas sin utilizar ambos. análisis de código estático y análisis dinámico.
A continuación se presentan tres razones clave por las que la validación de procesos es importante.
- Seguro de calidad. La validación de procesos ayuda a garantizar que los dispositivos médicos se fabriquen de manera consistente y confiable, lo que reduce el riesgo de defectos y garantiza un alto nivel de calidad.
- Seguridad del paciente. Al validar el proceso de fabricación, se pueden identificar y mitigar posibles riesgos y defectos, mejorando la seguridad del paciente.
- Cumplimiento de requisitos reglamentarios. Cumplir con los requisitos reglamentarios, como el Reglamento del sistema de calidad (QSR) de la FDA, requiere el uso de procesos validados. No hacerlo puede generar problemas de cumplimiento.
Requisitos reglamentarios y documento de orientación de la FDA
Los requisitos reglamentarios descritos en el Documento de orientación de la FDA sobre ciberseguridad de dispositivos médicos, están diseñados para abordar las crecientes preocupaciones con respecto a la seguridad de los dispositivos médicos en un entorno de atención médica cada vez más conectado y digital. A continuación se ofrece una explicación de los requisitos reglamentarios.
Autoridad legal
La FDA ahora tiene una nueva autoridad estatutaria en virtud de la Ley Ómnibus de Reforma de Alimentos y Medicamentos (FDORA), promulgada en diciembre de 2022. Esta ley permite a la FDA exigir información de ciberseguridad en las presentaciones de dispositivos médicos para “dispositivos cibernéticos” y exige que los fabricantes tomen medidas específicas. acciones para demostrar una seguridad razonable de que sus dispositivos y sistemas relacionados son “ciberseguros”. El incumplimiento de estos requisitos se considera un acto prohibido, sujeto a procesamiento.
Definición de “dispositivo cibernético”
El término "dispositivo cibernético" se define recientemente en la ley como un dispositivo que incluye software validado, instalado o autorizado por el patrocinador, puede conectarse a Internet y contiene características tecnológicas que podrían ser vulnerables a amenazas de ciberseguridad. Esta definición ayuda a aclarar qué dispositivos están sujetos a los nuevos requisitos de ciberseguridad.
Inclusión de información sobre ciberseguridad en las presentaciones previas a la comercialización
La nueva guía exige que los fabricantes incluyan información específica sobre ciberseguridad en sus presentaciones previas a la comercialización a la FDA. Estas presentaciones previas a la comercialización pueden incluir presentaciones de aprobación previa a la comercialización (PMA), 510(k) o de novo. La información de ciberseguridad es necesaria para garantizar que el dispositivo cumpla con los requisitos de ciberseguridad descritos en la ley.
Monitorear y abordar las vulnerabilidades de ciberseguridad posteriores a la comercialización
Los fabricantes de dispositivos deben presentar un plan para monitorear, identificar y abordar las vulnerabilidades y exploits de ciberseguridad posteriores a la comercialización. Este plan debe describir el enfoque del fabricante para monitorear y abordar continuamente los riesgos de ciberseguridad que pueden surgir después de que el dispositivo esté en el mercado.
Garantía razonable de ciberseguridad
Los fabricantes deben diseñar, desarrollar y mantener procesos y procedimientos para brindar una garantía razonable de que el dispositivo y sus sistemas relacionados son "ciberseguros". Este requisito enfatiza la necesidad de un enfoque proactivo para incorporar la seguridad en el diseño del dispositivo y mantenerla durante todo el ciclo de vida del dispositivo.
Lista de materiales del software
Se espera que los fabricantes proporcionen una lista de materiales de software (SBOM) como parte de sus envíos. Este proyecto de ley debe detallar los componentes de software comerciales, de código abierto y disponibles en el mercado utilizados en el dispositivo. Esta información es crucial para identificar y abordar posibles vulnerabilidades en la pila de software.
Cumplimiento de requisitos adicionales de la FDA
El documento de orientación también faculta a la FDA a establecer requisitos adicionales a través de regulaciones para demostrar que el dispositivo y los sistemas relacionados son ciberseguros. Los fabricantes deben estar preparados para cumplir con cualquier requisito adicional descrito por la FDA.
Acto prohibido por incumplimiento
Un aspecto clave de los requisitos reglamentarios es la inclusión de una nueva ley prohibida por ley. Esto significa que el incumplimiento de los requisitos de ciberseguridad de la FDA no es sólo una cuestión de cumplimiento sino también una violación legal. El gobierno tiene la autoridad para procesar penalmente las violaciones de estos requisitos o solicitar medidas cautelares contra las empresas que no los cumplan.
Cómo pueden cumplir los fabricantes de dispositivos médicos
Los fabricantes de dispositivos médicos deben cumplir con muchos requisitos para garantizar la seguridad, eficacia y calidad de sus productos. Exploraremos aspectos clave del cumplimiento de los requisitos reglamentarios de la FDA en dispositivos médicos, poniendo más énfasis en abordar los problemas de seguridad en los dispositivos médicos y diseñando controles y protocolos de validación para los fabricantes.
Abordar los problemas de seguridad en los dispositivos médicos
Para cumplir con las regulaciones de la FDA y priorizar la seguridad del paciente, los fabricantes de dispositivos médicos deben abordar las preocupaciones de seguridad de manera efectiva. A continuación se ofrecen algunos consejos para abordar los problemas de seguridad en los dispositivos médicos:
- Estandarice su código y realice una evaluación de riesgos. El primer paso para tener un dispositivo de software médico sin problemas de seguridad es escribir el software con reglas de codificación estandarizadas. Para el software ya existente, puede verificar y limpiar la base del código para asegurarse de que cumple con los estándares de codificación aceptables. Los fabricantes de dispositivos médicos pueden aprovechar las soluciones de análisis de código estático para probar y analizar el código fuente de sus programas de software. Además del análisis de código estático, otros tipos de pruebas de software que pueden ayudar a ofrecer un excelente software incluyen pruebas de integración, pruebas de seguridad, pruebas unitarias, pruebas de sistemas y pruebas de usabilidad. Limpiar la base del código del software y realizar análisis de riesgos ayuda a los fabricantes a identificar posibles vulnerabilidades de seguridad en sus dispositivos. Esto incluye evaluar los riesgos relacionados con violaciones de datos, acceso no autorizado y la integridad de la funcionalidad del dispositivo.
- Actualizaciones de software y gestión de parches. Actualice y parchee periódicamente el software del dispositivo para abordar las vulnerabilidades conocidas. Las actualizaciones oportunas ayudan a prevenir violaciones de seguridad que pueden aprovechar el software obsoleto.
- Almacenamiento seguro de datos. Asegúrese de que los datos del paciente se almacenen de forma segura, tanto en el dispositivo como durante la transmisión de datos. Utilice prácticas seguras de almacenamiento de datos y protocolos de cifrado recomendados por la industria para proteger la privacidad del paciente y cumplir con las normas de protección de datos.
- Colabora con expertos en ciberseguridad. Considere colaborar con expertos en ciberseguridad y profesionales que se especializan en seguridad de dispositivos médicos. Su experiencia puede ayudar a los fabricantes a identificar y mitigar posibles riesgos de seguridad.
Controles de diseño y protocolo de validación para fabricantes
Los controles de diseño y los protocolos de validación adecuados son componentes vitales para garantizar que los dispositivos médicos cumplan con los requisitos reglamentarios de la FDA. Estos controles ayudan a los fabricantes a desarrollar dispositivos seguros y eficaces que brinden los beneficios previstos a los pacientes.
A continuación se presentan consideraciones clave para implementar controles de diseño y protocolos de validación.
- Planificación del diseño y desarrollo. Los fabricantes deben establecer un plan claro de diseño y desarrollo que describa el alcance, los objetivos y las expectativas del dispositivo médico. Este plan debe incluir gestión de riesgos, estudios de usabilidad y cumplimiento normativo.
- Diseñar entradas y salidas. Defina entradas de diseño que especifiquen los requisitos del dispositivo y salidas de diseño que describan las características del dispositivo. Asegúrese de que las entradas de diseño se alineen con las necesidades del usuario y los requisitos reglamentarios.
- Verificación y validación. Implemente procesos rigurosos de verificación y validación para confirmar que el dispositivo cumple con sus requisitos de diseño y uso previsto. Esto incluye probar y evaluar el rendimiento y la seguridad del dispositivo.
- Archivo de historial de diseño (DHF). Mantenga un DHF integral que registre todas las actividades de diseño y desarrollo, incluida la documentación de los controles de diseño, los resultados de las pruebas y los cambios de diseño.
- Presentaciones regulatorias. Presentar notificaciones previas a la comercialización (510(k)), solicitudes De Novo o solicitudes de aprobación previa a la comercialización (PMA) precisas y completas es esencial para obtener la autorización o aprobación de la FDA para sus dispositivos médicos. Estas presentaciones brindan a la FDA la información necesaria sobre la seguridad y eficacia de su dispositivo, ayudándola a evaluar su idoneidad para el mercado. No completar estos envíos correctamente puede provocar retrasos o incluso el rechazo de su producto.
- Vigilancia poscomercialización (PMS). Establecer un sistema PMS es fundamental para monitorear e informar eventos adversos, quejas y mal funcionamiento de los dispositivos. El cumplimiento de los requisitos del PMS ayuda a identificar problemas de seguridad e implementar acciones correctivas rápidamente. Garantiza que los fabricantes supervisen activamente el rendimiento de sus dispositivos y aborden cualquier problema que surja del uso.
Herramientas y métodos para el cumplimiento de la FDA
Los fabricantes pueden emplear diversas herramientas y métodos para lograr y mantener el cumplimiento de las regulaciones de la FDA para dispositivos médicos. A continuación se analizan herramientas y métodos esenciales que pueden ayudar en la búsqueda del cumplimiento de la FDA.
Análisis estático para cumplir con los requisitos de la FDA
El análisis estático desempeña un papel fundamental para ayudar a los fabricantes de dispositivos médicos a cumplir los estrictos requisitos establecidos por la FDA.
Aquí hay cinco razones para adoptar el análisis estático para cumplir con los requisitos de la FDA.
- Detección temprana de problemas. Herramientas de análisis estático puede identificar problemas potenciales como errores de codificación, vulnerabilidades de seguridad y fallas de diseño en una etapa temprana del proceso de desarrollo. Esta detección temprana permite realizar correcciones oportunas, lo que reduce el riesgo de modificaciones costosas y que requieren mucho tiempo más adelante en el ciclo de desarrollo. Los estándares de codificación recomendados para C y C++ son MISRA C / C ++ y CERT C/C++.
- Calidad de software mejorada. El software de alta calidad es imperativo para la seguridad y eficacia de los dispositivos médicos. El análisis estático ayuda a los desarrolladores a crear código más limpio, más confiable y más seguro. Esto no sólo facilita el cumplimiento de la FDA, sino que también mitiga el riesgo de problemas posteriores a la comercialización y preocupaciones de seguridad.
- Cumplimiento de la ciberseguridad. Dada la creciente importancia de la ciberseguridad para los dispositivos médicos, la FDA pone gran énfasis en ella. Las herramientas de análisis estático pueden identificar debilidades y vulnerabilidades de seguridad en el software, garantizando la alineación con las directrices de la FDA sobre ciberseguridad.
- Documentación reglamentaria. El análisis estático puede proporcionar documentación e informes completos que demuestran el examen exhaustivo del software y los pasos tomados para abordar cualquier problema identificado. Esta documentación es esencial para las presentaciones e inspecciones de la FDA.
- Validación simplificada. Al identificar y abordar problemas potenciales de manera temprana, el análisis estático puede acelerar el proceso de validación, lo cual es un paso importante en el cumplimiento de la FDA. Esto ayuda a reducir los retrasos en la comercialización de dispositivos médicos.
Cómo garantizar una gestión adecuada de los riesgos
Dado el lugar que ocupa la gestión de riesgos en el logro de los objetivos o requisitos generales de la FDA, profundicemos en cómo se puede lograr una gestión de riesgos adecuada.
- Evaluación de riesgos. La evaluación de riesgos en la fabricación de dispositivos médicos es el paso inicial para identificar y comprender los peligros potenciales asociados con un dispositivo médico. Su propósito es evaluar sistemáticamente el diseño del dispositivo, el uso previsto, la biocompatibilidad y otros factores de riesgo para identificar riesgos potenciales. Un ejercicio de evaluación de riesgos normalmente implica formar un equipo multifuncional con experiencia diversa, incluidos ingenieros, médicos y especialistas en reglamentación.
- Análisis de riesgo. El análisis de riesgos tiene como objetivo evaluar y priorizar los riesgos identificados asignando una puntuación de riesgo basada en la probabilidad de ocurrencia y la gravedad del daño potencial. El análisis de riesgos implica cuantificar cada riesgo mediante la evaluación de su probabilidad y su impacto potencial. Dependiendo de su empresa, se puede utilizar una matriz de riesgos u otro sistema de puntuación para priorizar los riesgos. Se da prioridad a los elementos de alto riesgo para tomar medidas adicionales.
- Mitigación de riesgos. La mitigación de riesgos implica el desarrollo de estrategias para reducir o controlar los riesgos identificados a un nivel aceptable. Su objetivo es mejorar la seguridad y eficacia del dispositivo médico. Las estrategias de mitigación pueden adoptar diversas formas, incluidas modificaciones de diseño, características de seguridad, mejores materiales, mayor capacitación de los usuarios o mejores instrucciones de uso. El objetivo es abordar los riesgos identificados de manera que se minimice su impacto.
- Documentación. La documentación adecuada es esencial para la transparencia, la rendición de cuentas y el cumplimiento normativo. Proporciona un registro del proceso de gestión de riesgos y ayuda a demostrar la debida diligencia ante las autoridades reguladoras. Todos los aspectos del proceso de gestión de riesgos, incluidas las evaluaciones, análisis y planes de mitigación de riesgos, deben documentarse meticulosamente. Esta documentación se mantiene durante todo el ciclo de vida del producto.
- Mejora continua. El proceso de gestión de riesgos debe ser un sistema dinámico y en evolución. La mejora continua garantiza que sus prácticas de gestión de riesgos sigan siendo eficaces para abordar nuevos riesgos y condiciones cambiantes del mercado. Se realizan revisiones y actualizaciones periódicas para incorporar las lecciones aprendidas de proyectos anteriores y adaptarse a la evolución de la tecnología y las regulaciones. Esto puede implicar revisar y revisar las evaluaciones, análisis y planes de mitigación de riesgos.
Cómo el análisis estático para C/C++ puede ayudar con la gestión de riesgos
Muchos de nuestros clientes de dispositivos médicos, al comenzar desde cero, han tenido éxito al introducir análisis estático para C / C ++ siguiendo estos pasos:
- Revisar los lineamientos existentes en la organización. Incluso si están diseñados para aplicarse manualmente, asócielos tanto como sea posible con las fichas que se proporcionan con la herramienta de análisis estático. Una herramienta madura de análisis estático probablemente cubrirá la mayoría de ellos. Puede considerar crear verificadores personalizados para las pautas restantes que no se pudieron asignar a verificadores de análisis estáticos de inmediato.
- Revise los estándares de codificación populares, especialmente aquellos creados teniendo en cuenta la seguridad. Seleccione un subconjunto de pautas que su equipo debe seguir. Al seleccionar las pautas, tiene sentido seguir la categorización de los estándares y seleccionar las pautas que se clasifican como las más importantes. Por ejemplo, en CERT probablemente desee comenzar con las pautas L1 y en MISRA C 2023, querrá revisar las pautas obligatorias.
- Defina la configuración de la herramienta de análisis estático e incluya pautas específicas de su organización y pautas seleccionadas, como CERT. No habilite todas las fichas a la vez. En su lugar, comience con un pequeño subconjunto para evitar inundar a los desarrolladores con infracciones.
- Asegúrese de que sus desarrolladores puedan escanear su código inmediatamente después de su creación. También tiene sentido incluir análisis estático en el proceso de CI/CD.
- A medida que los desarrolladores avancen y limpien el código fuente, asegúrese de habilitar progresivamente más verificadores de su lista. Al final del día, lo que desea es proporcionar evidencia de que cumple con todo el conjunto de pautas que seleccionó, no de que se detuvo a la mitad. Para orientarse mejor en el proceso y comprender su progreso actual, puede implementar un sistema de informes central para ayudar a agregar los datos de prueba y monitorear el trabajo de los desarrolladores. Vea un ejemplo de esto a continuación:
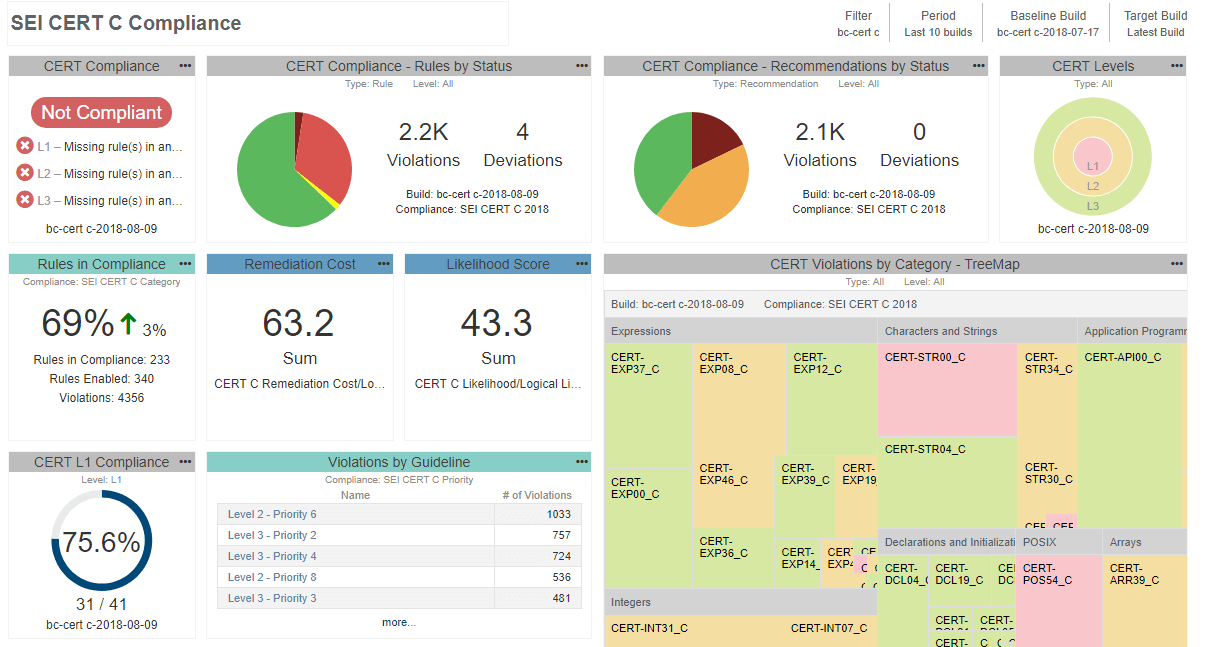
Dado que los informes de análisis estáticos pasan a formar parte del sistema de gestión de calidad, no se puede utilizar cualquier herramienta. La FDA exige que todas las herramientas utilizadas en el desarrollo y verificación del software estén validadas para el uso previsto. Hay diferentes formas de demostrar la idoneidad de la herramienta para su uso en desarrollos críticos para la seguridad. Dependiendo del riesgo del dispositivo, podría ser tan simple como reutilizar un certificado de cumplimiento o completar el proceso más largo de calificación de la herramienta.
Usando una certificación TÜV SÜD
Para el usuario final, la opción más conveniente es atribuirse el mérito del trabajo realizado por el proveedor de la herramienta y reutilizar la certificación otorgada para la herramienta de prueba por una organización de certificación externa como TÜV SÜD. Parasoft C / C ++test, por ejemplo, está cubierto con una certificación TÜV SÜD que se puede reutilizar para demostrar la idoneidad para desarrollar software de acuerdo con estándares médicos como IEC 62304.
Realización de la calificación de la herramienta
Para dispositivos de alto riesgo, como los de Clase C, es posible que necesite validar la herramienta internamente en su entorno de desarrollo. La intención es proporcionar evidencia de que la herramienta opera de acuerdo con sus requisitos operativos, recopilados en el entorno de desarrollo del proyecto. Este es un proceso muy tedioso y que requiere mucho tiempo.
La mejor situación es si su proveedor de herramientas puede apoyarlo en este esfuerzo y proporcionarle un kit de calificación de herramientas especial que contenga casos de prueba bien diseñados y el marco de automatización para ejecutarlos en el entorno de desarrollo del proyecto y generar automáticamente la documentación que pueda servir como la evidencia para la validación de la herramienta. Aquí nuevamente, la prueba C/C++ del producto estrella de Parasoft, proporciona una kit de calificación de herramientas automatizado.
Estudios de casos y aplicaciones del mundo real
Tenemos una gran cantidad de estudios de casos de aplicaciones del mundo real de las herramientas de análisis estático de Parasoft. Pero veamos dos historias de éxito distintas que ejemplifican cómo nuestra solución de análisis estático ha desempeñado un papel importante para ayudar a los fabricantes de dispositivos médicos a superar sus desafíos únicos y cumplir con los requisitos de la FDA.
Historias de éxito de fabricantes de dispositivos médicos
Los siguientes estudios de caso ilustran cómo las soluciones de prueba de software de Parasoft han sido fundamentales para ayudar a los fabricantes de dispositivos médicos a abordar sus desafíos únicos y alcanzar sus objetivos. Inovytec y Smiths Medical observaron mejoras notables en la calidad del código, el cumplimiento y la eficiencia de las pruebas a través de su colaboración con Parasoft.
El viaje de Inovytec hacia la certificación FDA 510(k)
Inovytec, una empresa dedicada a la producción de dispositivos médicos, se embarcó en la misión de lograr la certificación FDA 510(k) para su ventilador Ventway Sparrow. Su desafío consistía en ofrecer un código limpio y al mismo tiempo cumplir con las regulaciones de la FDA. La solución de análisis de código estático C/C++ de Parasoft vino a su rescate.
El equipo de desarrollo de software de Inovytec personalizó la prueba Parasoft C/C++ para alinearse con los estrictos requisitos de la FDA. Cada vez que se preparaban para lanzar una nueva versión de software, se aseguraban de que el análisis estático de Parasoft estuviera configurado para ejecutarse de acuerdo con las definiciones de las regulaciones de la FDA. El resultado no solo fue una mejor calidad del código, sino también un éxito rotundo al aprobar el 100 % de las normas y directrices de certificación FDA 510(k). Parasoft surgió como la solución de prueba preferida en Inovytec, y su colaboración con ESL, un distribuidor de productos Parasoft en Israel, brindó soporte y experiencia esenciales cuando fue necesario.
Adopción de Smiths Medical del desarrollo basado en pruebas (TDD)
Smiths Medical, un renombrado fabricante de dispositivos médicos especializados, enfrentó una serie de desafíos en su búsqueda por desarrollar dispositivos de alta calidad y críticos para la seguridad. software de dispositivos médicos. Las pruebas automatizadas desempeñan un papel crucial en la estrategia de pruebas de Smiths Medical.
Los intentos anteriores de incorporar herramientas no lograron un éxito total. El equipo de desarrollo buscó una solución que pudiera mejorar todo su proceso de prueba adoptando una nueva perspectiva centrada en las pruebas unitarias y el desarrollo basado en pruebas (TDD), una metodología que integra diseño, pruebas y desarrollo de código. Necesitaban una herramienta que pudiera encajar en su proceso de pruebas y mejorar su cultura de desarrollo general. La prueba Parasoft C/C++ demostró ser la respuesta a sus desafíos.
El equipo de software no solo logró la adopción exitosa de TDD, sino que también se benefició de una estabilidad de prueba mejorada, una cobertura de código mejorada y un proceso de calificación de herramientas simplificado, lo cual fue fundamental para las aplicaciones críticas para la seguridad. Con Parasoft, Smiths Medical pudo transformar sus procesos de desarrollo, haciendo de las pruebas una parte integral de su cartera de software y, en última instancia, garantizando la entrega de dispositivos médicos seguros y de alta calidad.
Superar los desafíos en la validación de la FDA
El proceso de validación de la FDA puede resultar complejo y desafiante para los fabricantes de dispositivos médicos. Sin embargo, hay una serie de medidas que los fabricantes pueden tomar para superar estos desafíos.
- Comenzar temprano. El proceso de validación de la FDA debería comenzar temprano en el ciclo de desarrollo del producto. Esto dará a los fabricantes más tiempo para planificar y ejecutar el proceso de validación y ayudará a identificar y abordar cualquier problema potencial desde el principio.
- Utilice un enfoque basado en el riesgo. El proceso de validación de la FDA debe basarse en una evaluación de riesgos del dispositivo. Los fabricantes deberían centrar sus esfuerzos de validación en los componentes y funciones más críticos del dispositivo.
- Utilice personal calificado. El proceso de validación de la FDA debe ser realizado por personal calificado que tenga experiencia con las regulaciones de la FDA y las mejores prácticas de validación.
- Documentar el proceso de validación. Los fabricantes deben documentar minuciosamente el proceso de validación de la FDA. Esto ayudará a los fabricantes a demostrar a la FDA que han validado su dispositivo de acuerdo con las regulaciones de la FDA.
Próximos pasos para navegar por la guía de validación de la FDA
La FDA proporciona una guía de validación esencial para que los fabricantes de dispositivos médicos garanticen la calidad del producto y la seguridad del paciente. A medida que las regulaciones y directrices evolucionan, es fundamental estar al tanto de estos cambios y planificar el cumplimiento futuro. Dos formas en que los fabricantes de dispositivos médicos pueden hacer esto es prepararse para futuras pautas de la FDA y seguir cumpliendo.
Preparación para futuras directrices de la FDA
La FDA actualiza y revisa constantemente sus documentos de orientación para los fabricantes de dispositivos médicos. Para prepararse para futuras directrices de la FDA, los fabricantes de dispositivos médicos deben:
- Supervise el sitio web de la FDA para obtener actualizaciones de los documentos de orientación. La FDA publica actualizaciones de sus documentos de orientación en su sitio web. Los fabricantes deben consultar periódicamente el sitio web de la FDA para obtener actualizaciones de los documentos de orientación que sean relevantes para sus dispositivos.
- Participe en talleres y seminarios web de la FDA. La FDA organiza con frecuencia talleres y seminarios web sobre una variedad de temas relacionados con la regulación de dispositivos médicos. Los fabricantes deben participar en estos eventos para mantenerse actualizados sobre las últimas ideas de la FDA sobre la regulación de dispositivos médicos.
- Establezca contactos con otros fabricantes de dispositivos médicos. Los fabricantes pueden establecer contactos con otros fabricantes de dispositivos médicos para compartir información y mejores prácticas para cumplir con las regulaciones de la FDA.
Garantizar el cumplimiento continuo
Los fabricantes de dispositivos médicos deben garantizar el cumplimiento continuo de las regulaciones de la FDA. Para ello, los fabricantes deberían:
- Establecer y mantener un sistema de calidad. Un sistema de calidad es un conjunto de procedimientos y procesos que los fabricantes utilizan para garantizar la seguridad y eficacia de sus dispositivos. Los fabricantes deben establecer y mantener un sistema de calidad que cumpla con los requisitos del Reglamento del sistema de calidad de la FDA.
- Realizar auditorías periódicas de su sistema de calidad. Los fabricantes deben realizar auditorías periódicas de su sistema de calidad para identificar y abordar cualquier área de incumplimiento.
- Presentar informes MDR a la FDA. Los fabricantes deben presentar informes de dispositivos médicos (MDR) a la FDA sobre los eventos adversos que ocurren con sus dispositivos. Al enviar informes MDR a la FDA, los fabricantes pueden ayudar a la FDA a identificar y abordar problemas de seguridad con sus dispositivos.
Con el tiempo, lograr el cumplimiento de la FDA ha demostrado ser bastante riguroso y requiere mucho tiempo. Sin embargo, con herramientas de análisis estático y dinámico, estos desafíos se pueden superar. Las herramientas de análisis estático ofrecen capacidades avanzadas de análisis de código estático, que ayudan a identificar y rectificar problemas en el software y el código en las primeras etapas del proceso de desarrollo.
La introducción del análisis estático es un esfuerzo dedicado que requiere tiempo y costos para el desarrollador. Pero es una forma comprobada de proteger su sistema contra ataques maliciosos. La implementación de análisis estático con un conjunto bien pensado de pautas de seguridad le permite crear sistemas que puedan resistir futuros ataques imprevistos.
Cumplimiento de software IEC 62304 en la industria médica
“MISRA”, “MISRA C” y el logotipo del triángulo son marcas comerciales registradas de The MISRA Consortium Limited. © The MISRA Consortium Limited, 2021. Todos los derechos reservados.



